The Genome in a Bottle Consortium (NIST)
We have collected the NIST Ashkenazim (HG003, HG004, HG002) Jewish data set for public use and comparison testing.
Quick data access:
- Project:
A1293-220310-VAR-NIST - Mosaic
- Ubox
- UCGD Data path:
/scratch/ucgd/lustre/UCGD_Datahub/IRBs/proj_UCGDCollab/A1293-220310-VAR-NIST/UCGD
Data collection.
The majority of WGS projects processed within the UCGD are ~30-50x coverage, so a subselection of the Illumina WGS 2x150bp 300X per individual data was collected. To adjust to the desired coverage levels, and based on this NIST README a single data directory was downloaded and processed for each individual, as opposed to downloaded the complete dataset and downsampling.
Important commands:
Initially the 140627_D00360_0030_AHA0L6ADXX directory was downloaded and processed but it was discovered that this directory had coverage of >1%, so the following directories were used.
Father:
$> aws s3 sync aws s3 sync s3://giab/data/AshkenazimTrio/HG003_NA24149_father/NIST_HiSeq_HG003_Homogeneity-12389378/HG003_HiSeq300x_fastq/140701_D00360_0032_AHA0KGADXX/ 140701_D00360_0032_AHA0KGADX/
mother:
$> aws s3 sync s3://giab/data/AshkenazimTrio/HG004_NA24143_mother/NIST_HiSeq_HG004_Homogeneity-14572558/HG004_HiSeq300x_fastq/140818_D00360_0046_AHA5R5ADXX/ 140818_D00360_0046_AHA5R5ADXX
son:
$> aws s3 sync s3://giab/data/AshkenazimTrio/HG002_NA24385_son/NIST_HiSeq_HG002_Homogeneity-10953946/HG002_HiSeq300x_fastq/140528_D00360_0018_AH8VC6ADXX/ 140528_D00360_0018_AH8VC6ADXX
Downloaded size comparison:
$> du -sch 140701_D00360_0032_AHA0KGADXX/
69G 140701_D00360_0032_AHA0KGADXX
$> du -sch 140818_D00360_0046_AHA5R5ADXX/
65G 140818_D00360_0046_AHA5R5ADXX/
$. du -sch 140528_D00360_0018_AH8VC6ADXX/
67G 140528_D00360_0018_AH8VC6ADXX
Project setup
Next the project was set up within UCGD using this papers summary information and this quick reference.
Comparison of version 2.0.0+ of the UCGD-Pipeline
A Benchmark comparison was run on individual HG002 (UCGD generated file) using the following steps and commands.
Downloaded benchmark files:
$> wget https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio/HG002_NA24385_son/latest/GRCh38/HG002_GRCh38_1_22_v4.2.1_benchmark.vcf.gz
$> wget https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio/HG002_NA24385_son/latest/GRCh38/HG002_GRCh38_1_22_v4.2.1_benchmark.vcf.gz.tbi
$> wget https://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/AshkenazimTrio/HG002_NA24385_son/latest/GRCh38/HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed
Isolated HG002 Individual
$> bcftools view -S HG002 A1293-220310-VAR-NIST_Final_1647108329.vcf.gz -O b -o HG002.vcf
## decomposed and sort HG002 individual
$> vt decompose HG002_UCGD.vcf.gz -o HG002_UCGD.decompose.vcf.gz
$> bcftools sort HG002_UCGD.decompose.vcf.gz -o HG002_UCGD.decompose.sort.vcf.gz
$> tabix -p vcf HG002_UCGD.decompose.sort.vcf.gz
Running RTG
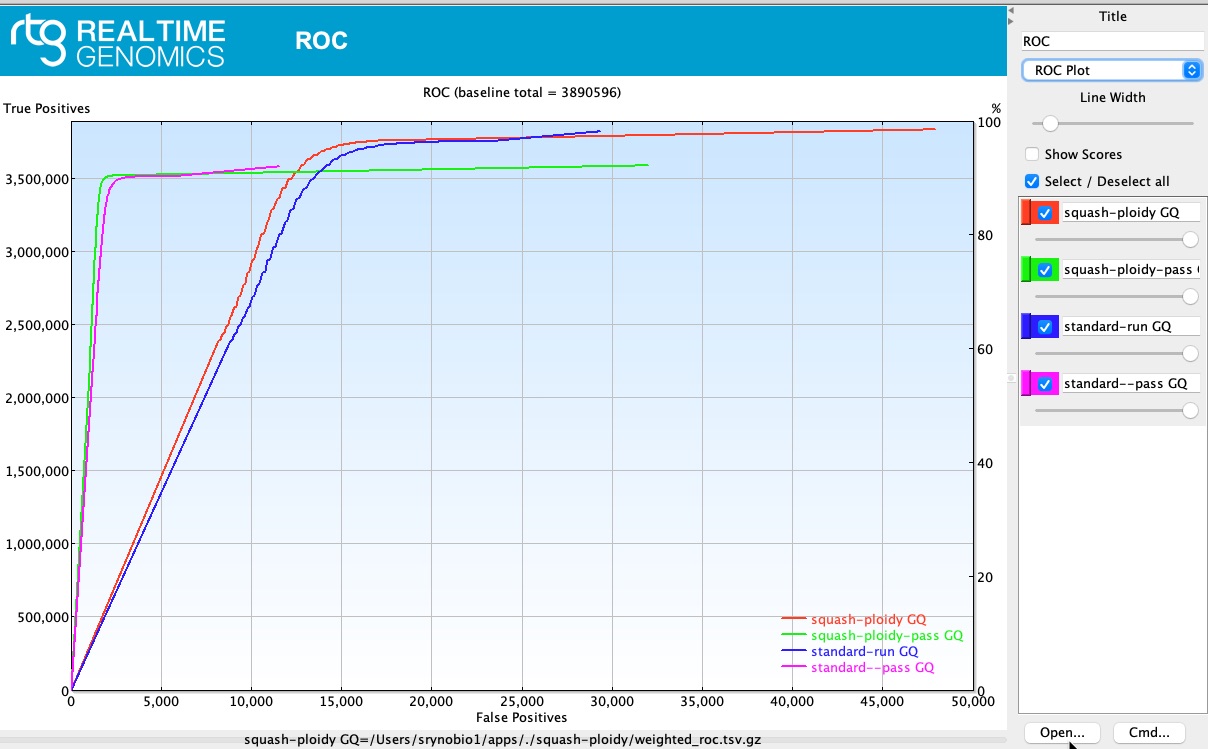
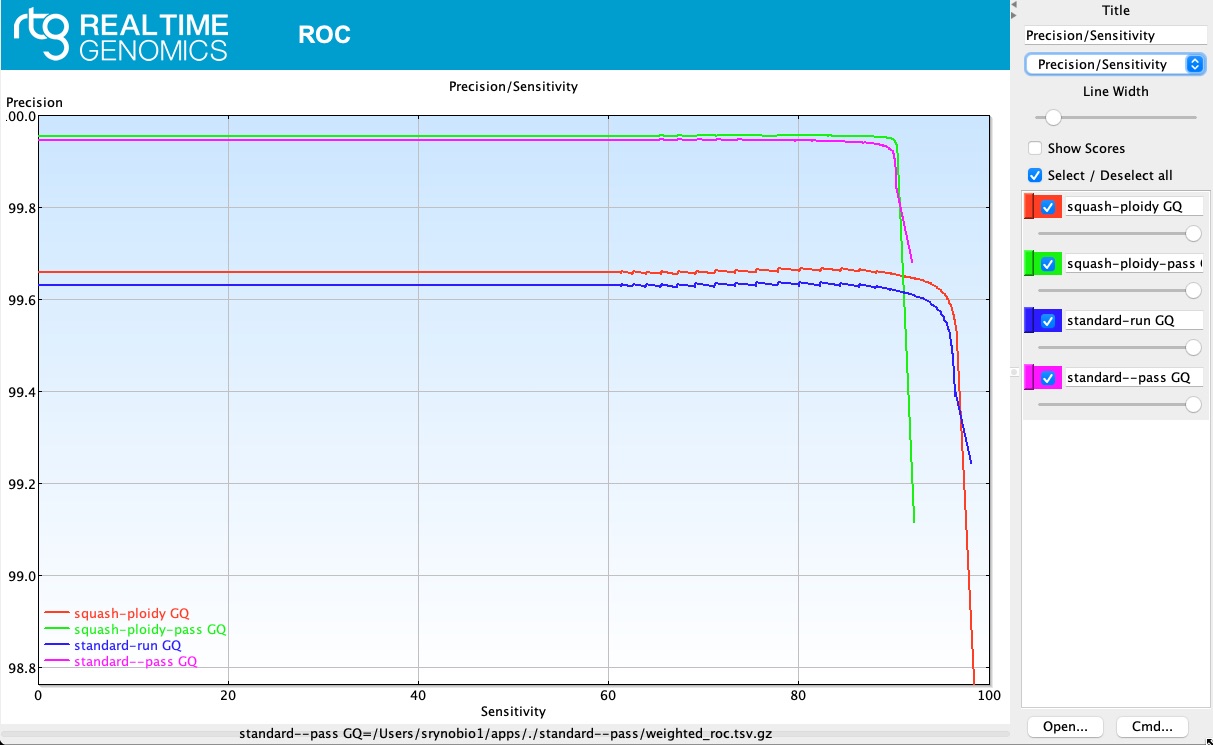
rtg vcfeval was used based on 4 different settings:
## (squash-ploidy GQ): Treat heterozygous genotypes as homozygous ALT in both baseline and calls, to allow matches that ignore zygosity differences.
## all-records: Allow use all records regardless of FILTER status
$> rtg-tools-3.12.1/rtg vcfeval -b HG002_GRCh38_1_22_v4.2.1_benchmark.sort.vcf.gz -c HG002_UCGD.decompose.sort.vcf.gz --bed-regions HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed --all-records --squash-ploidy -o squash-ploidy -t human_g1k_v38/
## (squash-ploidy-pass) squash-ploidy only allowing 'PASS' calls.
$> rtg-tools-3.12.1/rtg vcfeval -b HG002_GRCh38_1_22_v4.2.1_benchmark.sort.vcf.gz -c HG002_UCGD.decompose.sort.vcf.gz --bed-regions HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed --squash-ploidy -o squash-ploidy-pass -t human_g1k_v38/
## standard rtg vcfeval run with and without (--all-records)
## (standard-run)
$> rtg-tools-3.12.1/rtg vcfeval -b HG002_GRCh38_1_22_v4.2.1_benchmark.sort.vcf.gz -c HG002_UCGD.decompose.sort.vcf.gz --bed-regions HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed --all-records -o standard-run -t human_g1k_v38/
## (standard--pass)
$> rtg-tools-3.12.1/rtg vcfeval -b HG002_GRCh38_1_22_v4.2.1_benchmark.sort.vcf.gz -c HG002_UCGD.decompose.sort.vcf.gz --bed-regions HG002_GRCh38_1_22_v4.2.1_benchmark_noinconsistent.bed -o standard--pass -t human_g1k_v38
Results:
- Comparison done on Mar 21 2022
ROC
Precision/Sensitivity
Squash-ploidy-pass summary:
Overall Summary:
Threshold True-pos-baseline True-pos-call False-pos False-neg Precision Sensitivity F-measure
----------------------------------------------------------------------------------------------------
0.000 3516518 3516442 2515 374078 0.9993 0.9039 0.9492
None 3587391 3587305 32035 303205 0.9911 0.9221 0.955
Review of False Negative Calls:
To review the possible impact of false negative calls, VEP was ran on the rtg produced fn.vcf.gz file, and vep_annotation_reporter was used to collect ClinVar clinical significance annotations.
| Count | CLIN_SIG |
|---|---|
| 554 | benign |
| 63 | likely_benign |
| 18 | drug_response |
| 11 | uncertain_significance |
| 11 | protective |
| 8 | benign/likely_benign |
| 5 | pathogenic |
| 2 | conflicting_interpretations_of_pathogenicity |
| 1 | risk_factor |
Upon review of the pathogenic variants all were found to be co-annotated with the following terms:
benign
conflicting_interpretations_of_pathogenicity
uncertain_significance
Sentieon DNAscope comparison.
To be added when this comparison is complete.